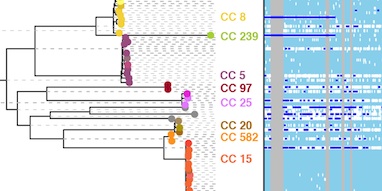
Two forms of phylogenetic distortion are caused by recombination. The first affects the shape of the tree topology. Although this is a potentially serious difficulty, Jessica Hedge and I recently showed that phylogenies estimated from whole bacterial genomes are surprisingly robust to this problem. The second affects the lengths of the branches. When genetic material is replaced by a homologous but distantly related sequence, it gives the appearance of a cluster of substitutions in the genome, and this can exaggerate branch lengths. ClonalFrameML detects these clusters of substitutions, identifies them as recombination events, and corrects the branch lengths of the tree.
Correcting for recombination is important in a variety of settings. In transmission studies, recent transmission between patients can be detected by comparing the genomes of the infecting bacteria. As we show in the paper, ClonalFrameML improves detection of transmission events by accounting for the tendency of recombination to elevate the evolutionary distance between genomes. We also report the discovery of a remarkably large chromosomal replacement event spanning 310 kilobases that may have led to the evolution of the ST582 strain of Staphylococcus aureus, underlining the importance of recombination over short and long timescales.
ClonalFrameML is a much faster implementation of the popular ClonalFrame method by Xavier and Daniel Falush. It is based on the same underlying assumptions and the same explicit evolutionary model, so it provides interpretable estimates of rates of recombination, the length of DNA imported by recombination, and the relative impact of recombination versus mutation. However, it can now analyse thousands of whole bacterial genomes in a matter of hours, representing a substantial improvement over the earlier method.
Thanks for the analysis of the accounting for recombination in bacterial phylogenies. I have learned a lot!
ReplyDeleteEmail me your assignments and I will send you back the solutions.
ReplyDeleteIn addition to solving problems I do projects, papers and essays.
I offer assistance in the following areas, undergraduate, graduate and post graduate levels:
Accounting
Accounting concept
Income statement
Balance sheet
Cash-flow statement
Accounts receivable, Note receivable
Inventory
Fixed Assets, Depreciation
Intangible assets
Current liability
Bond
Long-term liability
Common and preferred stock
Treasury stock
Cash and dividends
Financial statement analysis
Cost accounting, Cost analysis
Accounting and management decision
Budgeting
prashant8778@yahoo.co.in