This week sees publication of our paper
New Routes to Phylogeography: a Bayesian Structured Coalescent Approximation in
PLoS Genetics.
Phylogeography is the recovery of migration history from genome sequences, and has exploded as a field in recent years. Over a thousand papers have used contemporary sequences and ancient DNA to reconstruct migratory trends, locate the origin of outbreaks and track the spread of infectious diseases. In many high profile examples phylogeography has informed our understanding of how major human pathogens spread.
In our new paper we solve a severe and apparently widely unappreciated problem: that the most popular approaches to phylogeography are heavily biased, extremely sensitive to sampling structure and substantially underestimate statistical uncertainty. The problems stem from the treatment of migration as equivalent to mutation (discrete trait analysis; DTA), and the assumption that sampling locations are phylogeographically informative.
To solve these problems we introduce and demonstrate a new method
BASTA, implemented in the phylogenetic software package
BEAST2, that employs a novel approximation to enable inference under the structured coalescent – the bottom-up population genetics model of migration. Previously, methods for exact inference under the structured coalescent have proven too slow for many practical purposes, hence the need for a fast and accurate approximation.
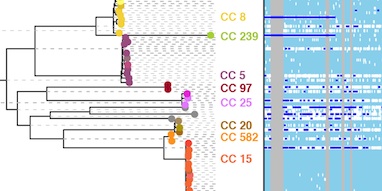
The biases we highlight with popular phylogeography methods are much more important than might appear from what is at one level a question of model choice. To underline this, we present an analysis of around 100 Ebola virus genome sequences to investigate the emergence of human outbreaks. Epidemiological studies have found that animals act as a reservoir, maintaining the virus between the sporadic human outbreaks that have unfolded over the past four decades, a scenario that our structured coalescent-based model correctly identifies.
Remarkably, DTA, the de facto standard method for phylogeography, wrongly concluded with high confidence that Ebola has been maintained since 1976 by undetected human-to-human transmission between outbreaks. Although such a conclusion would never be believed in the case of Ebola, it makes clear the potential for highly misleading inference about transmission that could, for much less well understood diseases, have serious implications for public health policy.
BASTA is the result of a lot of hard work by
Nicola De Maio, who is a James Martin Fellow at the Oxford Martin School
Institute for Emerging Infections, with help from Jessie Wu and
Kathleen O'Reilly. You can
read the paper here and
download BASTA here.