Thursday, 3 August 2017
New draft paper on combining p-values through the harmonic mean
In a preprint released today on Biorxiv I report a new method for improving the sensitivity to detect statistical signals by averaging over multiple alternative hypotheses using the harmonic mean p-value. The draft paper looks at example problems in genome-wide association studies (GWAS) in which signals of association may be apparent, but perhaps not sufficiently strong to meet the stringent threshold required to control for the millions of tests performed. Combining weak signals in arbitrary ways - for example across consecutive variants - can reveal signals sufficiently strong to meet the statistical significance threshold. This could be especially useful when looking for interactions, for example between host and pathogen genetics in their effect on infection, because it may be possible to conclude that a particular variant on the host side is involved, even if there is uncertainty over the specific pathogen variant it interacts with. Often such uncertainty arises because of the sheer number of possibilities. Similar ideas are beginning to gain traction in GWAS, and the ability to easily average over hypotheses is one of the strengths of Bayesian statistics. This new paper shows that the benefits of model averaging can be achieved easily in non-Bayesian statistics by taking the harmonic mean p-value from a range of tests. The test is very general and robust to a range of complexities including non-independence between the p-values.
Thursday, 29 September 2016
New paper: SCOTTI Efficient reconstruction of transmission within outbreaks with the structured coalescent

In this new paper Nicola De Maio, Jessie Wu and I introduce SCOTTI, a method for quickly and accurately inferring who-infected- whom from genomic and epidemiological data. SCOTTI addresses very widespread, but generally neglected problems in joint epidemiological and genomic inference, notably the presence of non-sampled and undetected intermediate cases and within-host pathogen variation caused by microevolution. Using real examples and simulations, we show that these problems cause strong misleading effects on existing popular inference methods. SCOTTI is based on BASTA, our recent breakthrough method for phylogeographic inference, and offers new standards of accuracy, calibration, and computational efficiency. SCOTTI is distributed as an open source package within BEAST2.
Friday, 23 September 2016
Prize PhD Studentships available
I am offering two PhD projects as part of the annual Nuffield Department of Medicine Prize Studentship competition:
In addition to my projects, the Modernising Medical Microbiology project has announced the following PhD projects as part of the competition:
- Real-time detection of multidrug resistant tuberculosis and transmission in England
Joint with David Wyllie, molecular microbiologist, this project is focused on developing statistical methods for recognizing transmission clusters, integrating genomics approaches with molecular typing schemes and developing future-proof taxonomy for strain identification. - Tracking future infection threats using genomic data and electronic health records
Joint with David Clifton, biomedical engineer, this project aims to develop new machine learning and statistical methods to identify genomic markers of antibiotic resistance and susceptibility within various pathogens, to help track future infection threats.
In addition to my projects, the Modernising Medical Microbiology project has announced the following PhD projects as part of the competition:
- Antimicrobial resistance gene/vector transmission across human, animal and environmental reservoirs
Supervised by Nicole Stoesser, Nicola De Maio and Derrick Crook - Healthcare big data and genomics for infectious disease threat detection
Supervised by David Clifton, David Eyre and Tim Peto - Prediction of Mycobacterium tuberculosis drug resistance through genome sequencing clinical samples
Supervised by Tim Walker and Tim Peto - Antibiotic resistance in Tuberculosis: Predicting de novo the effect of individual genetic mutations
Supervised by Phil Fowler and Sarah Walker
Friday, 19 August 2016
The Rsp virulence regulator: new review in Trends in Microbiology
In the September issue of Trends in Microbiology, Mark Smeltzer casts the spotlight on the story of rsp, a virulence regulator in Staphylococcus aureus that evolves within infected patients and may play a role in disease.
The new review covers recent work on the rsp gene including a series papers that my collaborators and my group have contributed:
Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation.
Das, S., Lindemann, C., Young, B. C., Muller, J., Österreich, B., Ternette, N., Winkler, A.-C., Paprotka, K., Reinhardt, R., Förstner, K. U., Allen, E., Flaxman, A., Yamaguchi, Y., Rollier, C. S., Van Diemen, P., Blättner, S., Remmele, C. W., Selle, M., Dittrich, M., Müller, T., Vogel, J., Ohlsen, K., Crook, D., Massey, R., Wilson, D. J., Rudel, T., Wyllie, D. H., and M. J. Fraunholz (2016)
Proceedings of the National Academy of Sciences USA 113: E3101–E3110. (abstract pdf)
Evolutionary trade-offs underlie the multi-faceted virulence of Staphylococcus aureus.
Laabei, M., Uhlemann, A.-C., Lowy, F. D., Austin, E. D., Yokoyama, M., Ouadi, K., Feil, E., Thorpe, H. A., Williams, B., Perkins, M., Peacock, S. J., Clarke, S. R., Dordel, J., Holden, M., Votintseva, A. A., Bowden, R., Crook, D. W., Young, B. C., Wilson, D. J., Recker, M. and R. C. Massey (2015)
PLoS Biology 13: e1002229. (abstract pdf)
Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease.
Young, B. C., Golubchik, T., Batty, E. M., Fung, R., Larner-Svennson, H., Votintseva, A., Miller, R. R., Godwin, H., Knox, K., Everitt, R. G., Iqbal, Z., Rimmer, A. J., Cule, M., Ip C. L. C., Didelot, X., Harding, R. M., Donnelly, P. J., Peto, T. E., Crook, D. W., Bowden, R. and D. J. Wilson (2012)
Proceedings of the National Academy of Sciences USA 109: 4550-4555. (abstract pdf F1000)
The new review covers recent work on the rsp gene including a series papers that my collaborators and my group have contributed:
Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation.
Das, S., Lindemann, C., Young, B. C., Muller, J., Österreich, B., Ternette, N., Winkler, A.-C., Paprotka, K., Reinhardt, R., Förstner, K. U., Allen, E., Flaxman, A., Yamaguchi, Y., Rollier, C. S., Van Diemen, P., Blättner, S., Remmele, C. W., Selle, M., Dittrich, M., Müller, T., Vogel, J., Ohlsen, K., Crook, D., Massey, R., Wilson, D. J., Rudel, T., Wyllie, D. H., and M. J. Fraunholz (2016)
Proceedings of the National Academy of Sciences USA 113: E3101–E3110. (abstract pdf)
Evolutionary trade-offs underlie the multi-faceted virulence of Staphylococcus aureus.
Laabei, M., Uhlemann, A.-C., Lowy, F. D., Austin, E. D., Yokoyama, M., Ouadi, K., Feil, E., Thorpe, H. A., Williams, B., Perkins, M., Peacock, S. J., Clarke, S. R., Dordel, J., Holden, M., Votintseva, A. A., Bowden, R., Crook, D. W., Young, B. C., Wilson, D. J., Recker, M. and R. C. Massey (2015)
PLoS Biology 13: e1002229. (abstract pdf)
Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease.
Young, B. C., Golubchik, T., Batty, E. M., Fung, R., Larner-Svennson, H., Votintseva, A., Miller, R. R., Godwin, H., Knox, K., Everitt, R. G., Iqbal, Z., Rimmer, A. J., Cule, M., Ip C. L. C., Didelot, X., Harding, R. M., Donnelly, P. J., Peto, T. E., Crook, D. W., Bowden, R. and D. J. Wilson (2012)
Proceedings of the National Academy of Sciences USA 109: 4550-4555. (abstract pdf F1000)
Wednesday, 3 August 2016
Thursday, 30 June 2016
Friday, 17 June 2016
Collaborative PhD and postdoc positions available
Dr Nicole Stoesser, Prof. Derrick Crook, myself and colleagues in Oxford are seeking a postdoc in Microbial Genomics with statistics skills to join a new three-year project investigating antimicrobial resistance in environmental, human and animal reservoirs of E. coli and related organisms. The application deadline is noon Monday 11th July. For more details click here.
Dr Pierre Mahe of bioMérieux in Grenoble, France, is seeking to appoint an industry-linked PhD position developing statistical methods for genome-based characterization of antimicrobial resistance and virulence genes, with a focus on the opportunistic pathogen Pseudomonas aeruginosa. The position involves a secondment here in Oxford. For more details click here or contact Pierre Mahe.
Dr Pierre Mahe of bioMérieux in Grenoble, France, is seeking to appoint an industry-linked PhD position developing statistical methods for genome-based characterization of antimicrobial resistance and virulence genes, with a focus on the opportunistic pathogen Pseudomonas aeruginosa. The position involves a secondment here in Oxford. For more details click here or contact Pierre Mahe.
Tuesday, 17 May 2016
New paper: How low-toxic Staph. aureus mutants cause severe infections

reduce toxicity while maintaining the ability to survive, proliferate and cause infection within the human body.
In previous work, we have found that Staph. aureus evolves by mutation within the body quickly enough to influence the progression of disease, and that diversity generated by evolution in the body is a widespread phenomenon. In the case of one patient who we followed longitudinally for over a year, we identified that bacteria in the bloodstream differed from those in the nose by several mutations, of which a loss-of-function mutation in the rsp regulatory gene represented the most likely candidate for playing a possible role in causing severe infection.
We collaborated with Ruth Massey at Bath who discovered to our surprise that while rsp loss-of-function mutants do indeed show differences in toxicity - one of several traditional correlates of virulence readily measured in the laboratory - they showed reduced toxicity. Going further, Ruth and her collaborators showed that bloodstream infections in general show reduced toxicity compared to milder skin infections and asymptomatically carried nose populations, overturning previous views on the relationship between Staph. aureus toxicity and virulence.
Today's new paper offers a detailed dissection of rsp. Working with Claudia Lindemann and David Wyllie at the University of Oxford and Martin Fraunholz and collaborators at the University of Würzburg, we found that although rsp mutants show reduced toxicity, crucially they retain their capacity to survive, grow, spread through the body and cause abscesses. In other words, rsp uncouples toxicity from pathogenicity. This decoupling could be important for evading the immune system and establishing severe infections. To find out more, see the full paper.
Tuesday, 12 April 2016
Postdoctoral Scientist in Statistical Genomics
We are recruiting for a Postdoctoral Scientist in Statistical Genomics
working on Antimicrobial Resistance (AMR) gene discovery and focused on
Tuberculosis. This will be a joint position at the University of Oxford between Derrick Crook's group and mine, and part of the large international CRyPTIC consortium.
The role is for a population geneticist or statistical geneticist to develop and apply statistical methods, including genome-wide association studies, for discovering rare and common genetic variants underlying antimicrobial resistance in Mycobacterium tuberculosis.
One third of the world's population - 2.5 billion people - are thought to be infected with tuberculosis (TB). This post offers an opportunity to work with global TB experts from five continents, statistical geneticists, clinicians, medical statisticians and software engineers; integrating statistical genetics, bioinformatics and machine learning methods with the aim of uncovering all genomic variants causing at least 1% resistance to first line anti-TB drugs.
We're looking for candidates with a PhD in genomics, evolutionary biology, statistics or a related subject. The post is full-time and fixed-term for up to 3 years initially.
The deadline for applications is noon on Friday 6th May 2016.
The role is for a population geneticist or statistical geneticist to develop and apply statistical methods, including genome-wide association studies, for discovering rare and common genetic variants underlying antimicrobial resistance in Mycobacterium tuberculosis.
One third of the world's population - 2.5 billion people - are thought to be infected with tuberculosis (TB). This post offers an opportunity to work with global TB experts from five continents, statistical geneticists, clinicians, medical statisticians and software engineers; integrating statistical genetics, bioinformatics and machine learning methods with the aim of uncovering all genomic variants causing at least 1% resistance to first line anti-TB drugs.
We're looking for candidates with a PhD in genomics, evolutionary biology, statistics or a related subject. The post is full-time and fixed-term for up to 3 years initially.
The deadline for applications is noon on Friday 6th May 2016.
Thursday, 7 April 2016
Making the most of bacterial GWAS: new paper in Nature Microbiology
In a new paper published this week in Nature Microbiology, we report the performance of genome wide association studies (GWAS) in bacteria to identify causal mechanisms of antibiotic resistance in four major pathogens, and introduce a new method, bugwas, to make the most of bacterial GWAS for traits under less strong selection.
As explained by Sarah Earle, joint first author with Jessie Wu and Jane Charlesworth, the problem with GWAS in bacteria is strong population structure and the consequent strong coinheritance of genetic variants throughout the genome. This phenomenon - known as genome-wide linkage disequilibrium (LD) - comes about because exchange of genes is relatively infrequent in bacteria, which reproduce clonally, compared to organisms that exchange genes every generation through sexual reproduction.
Genome-wide LD makes it difficult for GWAS to distinguish variants that causally influence a trait from other, coinherited variants that have no direct effect on the trait.
In the case of antibiotic resistance - a trait of high importance to human health - bacteria are under extraordinary selection pressures because resistance is a matter of life and death, to them as well as their human host. This helps overcome coinheritance and pinpoint causal variants because antibiotic usage selects for the independent evolution of the same resistance-causing variants in different genetic backgrounds.
Consequently, bacterial GWAS works very efficiently for antibiotic resistance: the variants most significantly associated with antibiotic resistance in 26 out of the 27 GWAS we performed were genuine resistance-conferring mutations. In the 27th we uncovered a putative novel mechanism of resistance to cefazolin in E. coli. These results for 17 antibiotics (ampicillin, cefazolin, cefuroxime, ceftriaxone, ciprofloxacin, erythromycin, ethambutol, fusidic acid, gentamicin, isoniazid, penicillin, pyrazinamide, methicillin, rifampicin, tetracycline, tobramycin and trimethoprim) across four species (E. coli, K. pneumoniae, M. tuberculosis and S. aureus) build on earlier work investigating beta-lactam resistance in S. pneumoniae, and convincingly demonstrate the potential for bacterial GWAS to discover new genes underlying important traits under strong selection.
What about traits under less strong selection, which probably includes pretty much every other bacterial trait? We show in this context that coinheritance poses a major challenge, based on detailed simulations. Often it may not be possible to use GWAS to pinpoint individual variants responsible for different traits because they are coinherited with - possibly many - other uninvolved variants.
But all is not lost. We show that even when individual locus-level effects cannot be pinpointed, there is often excellent power to characterize lineage-level differences in phenotype between strains. This is helpful for multiple reasons: (1) we often conceptualize trait variability in bacteria at the level of strain-to-strain differences (2) these differences can be highly predictive (3) we can prioritize variants for functional follow-up based on their contribution to strain-level differences.
These concepts represent a substantial departure from regular GWAS. In the human setting for instance, lineage-level differences are usually discarded as uninteresting or artefactual, and variants are almost always prioritized based on statistical evidence for involvement over-and-above any contribution to lineage-level differences. In the bacterial setting, we are forced to depart from these conventions because a large proportion of all genetic variation is strongly strain-stratified. To find out more, see the paper and try our methods.
As explained by Sarah Earle, joint first author with Jessie Wu and Jane Charlesworth, the problem with GWAS in bacteria is strong population structure and the consequent strong coinheritance of genetic variants throughout the genome. This phenomenon - known as genome-wide linkage disequilibrium (LD) - comes about because exchange of genes is relatively infrequent in bacteria, which reproduce clonally, compared to organisms that exchange genes every generation through sexual reproduction.
Genome-wide LD makes it difficult for GWAS to distinguish variants that causally influence a trait from other, coinherited variants that have no direct effect on the trait.
In the case of antibiotic resistance - a trait of high importance to human health - bacteria are under extraordinary selection pressures because resistance is a matter of life and death, to them as well as their human host. This helps overcome coinheritance and pinpoint causal variants because antibiotic usage selects for the independent evolution of the same resistance-causing variants in different genetic backgrounds.
Consequently, bacterial GWAS works very efficiently for antibiotic resistance: the variants most significantly associated with antibiotic resistance in 26 out of the 27 GWAS we performed were genuine resistance-conferring mutations. In the 27th we uncovered a putative novel mechanism of resistance to cefazolin in E. coli. These results for 17 antibiotics (ampicillin, cefazolin, cefuroxime, ceftriaxone, ciprofloxacin, erythromycin, ethambutol, fusidic acid, gentamicin, isoniazid, penicillin, pyrazinamide, methicillin, rifampicin, tetracycline, tobramycin and trimethoprim) across four species (E. coli, K. pneumoniae, M. tuberculosis and S. aureus) build on earlier work investigating beta-lactam resistance in S. pneumoniae, and convincingly demonstrate the potential for bacterial GWAS to discover new genes underlying important traits under strong selection.
What about traits under less strong selection, which probably includes pretty much every other bacterial trait? We show in this context that coinheritance poses a major challenge, based on detailed simulations. Often it may not be possible to use GWAS to pinpoint individual variants responsible for different traits because they are coinherited with - possibly many - other uninvolved variants.
But all is not lost. We show that even when individual locus-level effects cannot be pinpointed, there is often excellent power to characterize lineage-level differences in phenotype between strains. This is helpful for multiple reasons: (1) we often conceptualize trait variability in bacteria at the level of strain-to-strain differences (2) these differences can be highly predictive (3) we can prioritize variants for functional follow-up based on their contribution to strain-level differences.
These concepts represent a substantial departure from regular GWAS. In the human setting for instance, lineage-level differences are usually discarded as uninteresting or artefactual, and variants are almost always prioritized based on statistical evidence for involvement over-and-above any contribution to lineage-level differences. In the bacterial setting, we are forced to depart from these conventions because a large proportion of all genetic variation is strongly strain-stratified. To find out more, see the paper and try our methods.
Wednesday, 30 March 2016
CRyPTIC: rapid diagnosis of drug resistance in TB
The Modernising Medical Microbiology consortium has announced a new worldwide collaboration called CRyPTIC to speed up diagnosis of antibiotic resistant tuberculosis (TB).
TB infects nearly 10 million people each year and kills 1.5 million, making it one of the leading causes of death worldwide. Almost half a million people each year develop multidrug-resistant (MDR) TB, which defies common TB treatments. Time consuming tests must be run to identify MDR-TB and which drugs will work or fail. This delays diagnosis and creates uncertainty about the best drugs to prescribe to individual patients.
CRyPTIC aims to hasten the identification of MDR-TB using whole genome sequencing to identify genetic variants that give resistance to particular drugs. The project is funded by a $2.2m grant from the Bill & Melinda Gates Foundation and a £4m grant from the Wellcome Trust and MRC Newton Fund.
CRyPTIC aims to collect and analyse 100,000 TB cases from across the world, providing a database of MDR-TB that will underpin diagnosis using WGS. Samples from across Africa, Asia, Europe and the Americas will be collected by teams at more than a dozen centres They will conduct drug resistance testing and much of the genome sequencing. Read more information here.
TB infects nearly 10 million people each year and kills 1.5 million, making it one of the leading causes of death worldwide. Almost half a million people each year develop multidrug-resistant (MDR) TB, which defies common TB treatments. Time consuming tests must be run to identify MDR-TB and which drugs will work or fail. This delays diagnosis and creates uncertainty about the best drugs to prescribe to individual patients.
CRyPTIC aims to hasten the identification of MDR-TB using whole genome sequencing to identify genetic variants that give resistance to particular drugs. The project is funded by a $2.2m grant from the Bill & Melinda Gates Foundation and a £4m grant from the Wellcome Trust and MRC Newton Fund.
CRyPTIC aims to collect and analyse 100,000 TB cases from across the world, providing a database of MDR-TB that will underpin diagnosis using WGS. Samples from across Africa, Asia, Europe and the Americas will be collected by teams at more than a dozen centres They will conduct drug resistance testing and much of the genome sequencing. Read more information here.
Saturday, 5 March 2016
Snow Monkeys in Japan
Recently got back from the SMBE Satellite meeting on Pathogen Genomics in Japan. The organizers did a fantastic job and the talks were great. There was also time to visit the Japanese macaques at Snow Monkey Park, where one of the little guys climbed on to my shoulders
Thanks Ashlee Earl for the video and Koji Yahara, Alan McNally and Nick Croucher for additional commentary!
Wednesday, 20 January 2016
Nature Reviews Microbiology: Within-host evolution of bacterial pathogens
 Our new review of what genomics has taught us about Within-host evolution of bacterial pathogens has been published in Nature Reviews Microbiology.
Our new review of what genomics has taught us about Within-host evolution of bacterial pathogens has been published in Nature Reviews Microbiology.Friday, 9 October 2015
PLoS Biology: Staphylococcus aureus invading the blood are less toxic
 |
| Toxicity in nose, blood and skin bacteria. |
The notion that isolates responsible for serious human infection are less toxic challenges some long-held beliefs about the mechanism of disease in Staphylococcus aureus infections. Most models of disease assume a straightforward relationship between increased toxicity and greater virulence - the propensity to cause, or severity of, disease.
To test her observation, Ruth collaborated with groups from New York and Cambridge to investigate whether the pattern observed in one patient held more generally across 134 Staphylococcus aureus belonging to the notorious USA300 strain. It did.
Curiously, bacteria isolated from the skin and from superficial infections were equally toxic to nose bacteria. These findings raise new questions about the role of toxicity in colonization, transmission and serious infections of Staphylococcus aureus. One possibility that we wish to investigate further is whether toxicity might be required for the usual transmission of Staphylococcus aureus populations in the nose, skin or superficial infections (such as impetigo), whereas loss of toxicity may promote transition to deep tissue and bloodstream infections by evading immune defences.
Tuesday, 8 September 2015
New paper: Rapid host switching in Campylobacter
Our new open access paper Rapid host switching in generalist Campylobacter strains erodes the signal for tracing human infections was published last week in the ISME Journal.
 With Bethany Dearlove, Sam Sheppard and colleagues, we investigated common strains of campylobacter, the most frequent cause of bacterial gastroenteritis worldwide. Campylobacter infection is associated with food poisoning, particularly contaminated chicken. But in previous work, we found that certain strains (the ST-21, ST-45 and ST-828 complexes) are often found contaminating a range of meat and poultry, making it difficult to trace the source of human infection.
With Bethany Dearlove, Sam Sheppard and colleagues, we investigated common strains of campylobacter, the most frequent cause of bacterial gastroenteritis worldwide. Campylobacter infection is associated with food poisoning, particularly contaminated chicken. But in previous work, we found that certain strains (the ST-21, ST-45 and ST-828 complexes) are often found contaminating a range of meat and poultry, making it difficult to trace the source of human infection.
That previous work was based on partial genome sequencing known as MLST. In MLST, less than 1% of the information in the genome is captured. Now that whole genome sequencing is available, the expectation was that we should be able to distinguish easily between between ST-21, 45 and 828 strains contaminating poultry versus beef versus lamb, and so on.
What we found was surprising. Instead of these strains harbouring previously unobserved sub-structure that allowed them to be associated with different animal sources, we found rapidly mixing populations undergoing extremely fast transmission between animal species, with campylobacter strains ricocheting among animal species on a timescale of just a few years. This is faster than they can accumulate enough mutations to differentiate populations colonizing different animal species.
Our results present an unforeseen roadblock to tracing transmission with whole genome sequencing, and suggests these strains are adapted to a generalist lifestyle, shedding new light on the ecology of this pathogen. These findings push back against the tide of opinion that whole genome sequencing is necessarily a panacea for detecting transmission, and demonstrate that going forwards, a detailed understanding of the biology of zoonotic bacteria (those transmitting between multiple species) and intensive sampling of potential sources are essential for effectively tracing the source of human infection.
That previous work was based on partial genome sequencing known as MLST. In MLST, less than 1% of the information in the genome is captured. Now that whole genome sequencing is available, the expectation was that we should be able to distinguish easily between between ST-21, 45 and 828 strains contaminating poultry versus beef versus lamb, and so on.
What we found was surprising. Instead of these strains harbouring previously unobserved sub-structure that allowed them to be associated with different animal sources, we found rapidly mixing populations undergoing extremely fast transmission between animal species, with campylobacter strains ricocheting among animal species on a timescale of just a few years. This is faster than they can accumulate enough mutations to differentiate populations colonizing different animal species.
Our results present an unforeseen roadblock to tracing transmission with whole genome sequencing, and suggests these strains are adapted to a generalist lifestyle, shedding new light on the ecology of this pathogen. These findings push back against the tide of opinion that whole genome sequencing is necessarily a panacea for detecting transmission, and demonstrate that going forwards, a detailed understanding of the biology of zoonotic bacteria (those transmitting between multiple species) and intensive sampling of potential sources are essential for effectively tracing the source of human infection.
Monday, 17 August 2015
BASTA: Improved method for phylogeography
This week sees publication of our paper New Routes to Phylogeography: a Bayesian Structured Coalescent Approximation in PLoS Genetics.
Phylogeography is the recovery of migration history from genome sequences, and has exploded as a field in recent years. Over a thousand papers have used contemporary sequences and ancient DNA to reconstruct migratory trends, locate the origin of outbreaks and track the spread of infectious diseases. In many high profile examples phylogeography has informed our understanding of how major human pathogens spread.
In our new paper we solve a severe and apparently widely unappreciated problem: that the most popular approaches to phylogeography are heavily biased, extremely sensitive to sampling structure and substantially underestimate statistical uncertainty. The problems stem from the treatment of migration as equivalent to mutation (discrete trait analysis; DTA), and the assumption that sampling locations are phylogeographically informative.
 To solve these problems we introduce and demonstrate a new method BASTA, implemented in the phylogenetic software package BEAST2, that employs a novel approximation to enable inference under the structured coalescent – the bottom-up population genetics model of migration. Previously, methods for exact inference under the structured coalescent have proven too slow for many practical purposes, hence the need for a fast and accurate approximation.
To solve these problems we introduce and demonstrate a new method BASTA, implemented in the phylogenetic software package BEAST2, that employs a novel approximation to enable inference under the structured coalescent – the bottom-up population genetics model of migration. Previously, methods for exact inference under the structured coalescent have proven too slow for many practical purposes, hence the need for a fast and accurate approximation.
The biases we highlight with popular phylogeography methods are much more important than might appear from what is at one level a question of model choice. To underline this, we present an analysis of around 100 Ebola virus genome sequences to investigate the emergence of human outbreaks. Epidemiological studies have found that animals act as a reservoir, maintaining the virus between the sporadic human outbreaks that have unfolded over the past four decades, a scenario that our structured coalescent-based model correctly identifies.
Remarkably, DTA, the de facto standard method for phylogeography, wrongly concluded with high confidence that Ebola has been maintained since 1976 by undetected human-to-human transmission between outbreaks. Although such a conclusion would never be believed in the case of Ebola, it makes clear the potential for highly misleading inference about transmission that could, for much less well understood diseases, have serious implications for public health policy.
BASTA is the result of a lot of hard work by Nicola De Maio, who is a James Martin Fellow at the Oxford Martin School Institute for Emerging Infections, with help from Jessie Wu and Kathleen O'Reilly. You can read the paper here and download BASTA here.
Phylogeography is the recovery of migration history from genome sequences, and has exploded as a field in recent years. Over a thousand papers have used contemporary sequences and ancient DNA to reconstruct migratory trends, locate the origin of outbreaks and track the spread of infectious diseases. In many high profile examples phylogeography has informed our understanding of how major human pathogens spread.
In our new paper we solve a severe and apparently widely unappreciated problem: that the most popular approaches to phylogeography are heavily biased, extremely sensitive to sampling structure and substantially underestimate statistical uncertainty. The problems stem from the treatment of migration as equivalent to mutation (discrete trait analysis; DTA), and the assumption that sampling locations are phylogeographically informative.
The biases we highlight with popular phylogeography methods are much more important than might appear from what is at one level a question of model choice. To underline this, we present an analysis of around 100 Ebola virus genome sequences to investigate the emergence of human outbreaks. Epidemiological studies have found that animals act as a reservoir, maintaining the virus between the sporadic human outbreaks that have unfolded over the past four decades, a scenario that our structured coalescent-based model correctly identifies.
Remarkably, DTA, the de facto standard method for phylogeography, wrongly concluded with high confidence that Ebola has been maintained since 1976 by undetected human-to-human transmission between outbreaks. Although such a conclusion would never be believed in the case of Ebola, it makes clear the potential for highly misleading inference about transmission that could, for much less well understood diseases, have serious implications for public health policy.
BASTA is the result of a lot of hard work by Nicola De Maio, who is a James Martin Fellow at the Oxford Martin School Institute for Emerging Infections, with help from Jessie Wu and Kathleen O'Reilly. You can read the paper here and download BASTA here.
Friday, 24 July 2015
New Journal: Microbial Genomics

"Microbial Genomics (MGen) publishes high quality, original research on archaea, bacteria, microbial eukaryotes and viruses. MGen welcomes papers that use genomic approaches to understand microbial evolution, population genomics and phylogeography, outbreaks and epidemiological investigations, impact of climate or changing niche, metagenomic and whole transcriptome studies, and bioinformatic analysis covering the breadth of microbiology, from clinically important pathogens to microbial life in diverse ecosystems."
The journal, whose tag line is Bases to Biology, will publish microbiological discoveries and innovations in research methods and bioinformatics. The journal is headed by renowned Wellcome Trust Sanger Institute scientists Stephen Bentley and Nicholas Thompson with an impressive editorial board that I joined earlier this year. Article processing charges have been waived during the journal's launch year - so get in there fast!
Tuesday, 21 July 2015
Resistance is Futile: Science Museum Lates and Cheltenham Science Festival
Some photos from this summer's Science Museum Lates event with the Royal Society and the Cheltenham Science Festival. Thanks to everyone who helped: Liz Batty, Phelim Bradley, Jane Charlesworth, Dilly De Silva, Sarah Earle, Nicki Fawcett, Jess Hedge, Brian Mackenwells, Amy Mason, Charvy Narain, Anna Sheppard and Jessie Wu!


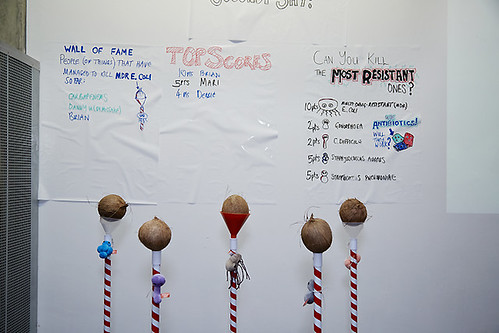

We had two activities. Dance Dance Evolution is a computer game which uses an adapted dance-dance mat with four squares representing bases in the DNA (A, C, G and T). Participants act as the DNA replicator, and mistakes cause mutations in the DNA sequence. The next dancer copies the sequence left by the previous dancer, demonstrating evolution by mutation over time. The game shows the percentage similarity of the current sequence to the original sequence, showing the amount of 'evolution' over the time period of the game. We discussed with visitors the relevance of this to the development of antibiotic resistance.


We had two activities. Dance Dance Evolution is a computer game which uses an adapted dance-dance mat with four squares representing bases in the DNA (A, C, G and T). Participants act as the DNA replicator, and mistakes cause mutations in the DNA sequence. The next dancer copies the sequence left by the previous dancer, demonstrating evolution by mutation over time. The game shows the percentage similarity of the current sequence to the original sequence, showing the amount of 'evolution' over the time period of the game. We discussed with visitors the relevance of this to the development of antibiotic resistance.
Wednesday, 8 April 2015
World Health Day: Food-borne disease theme
For World Health Day 2015, the group's research into food-borne campylobacter infection was featured on the Nuffield Department of Medicine's home page. The piece features recent work Bethany Dearlove and I have conducted into zoonotic (animal-human) transmission with Sam Sheppard. The paper is currently under review, and a preprint can be downloaded from the website.
Tuesday, 31 March 2015
ClonalFrameML: accounting for recombination in bacterial phylogenies
Two forms of phylogenetic distortion are caused by recombination. The first affects the shape of the tree topology. Although this is a potentially serious difficulty, Jessica Hedge and I recently showed that phylogenies estimated from whole bacterial genomes are surprisingly robust to this problem. The second affects the lengths of the branches. When genetic material is replaced by a homologous but distantly related sequence, it gives the appearance of a cluster of substitutions in the genome, and this can exaggerate branch lengths. ClonalFrameML detects these clusters of substitutions, identifies them as recombination events, and corrects the branch lengths of the tree.
Correcting for recombination is important in a variety of settings. In transmission studies, recent transmission between patients can be detected by comparing the genomes of the infecting bacteria. As we show in the paper, ClonalFrameML improves detection of transmission events by accounting for the tendency of recombination to elevate the evolutionary distance between genomes. We also report the discovery of a remarkably large chromosomal replacement event spanning 310 kilobases that may have led to the evolution of the ST582 strain of Staphylococcus aureus, underlining the importance of recombination over short and long timescales.
ClonalFrameML is a much faster implementation of the popular ClonalFrame method by Xavier and Daniel Falush. It is based on the same underlying assumptions and the same explicit evolutionary model, so it provides interpretable estimates of rates of recombination, the length of DNA imported by recombination, and the relative impact of recombination versus mutation. However, it can now analyse thousands of whole bacterial genomes in a matter of hours, representing a substantial improvement over the earlier method.
Friday, 28 November 2014
New paper: bacterial phylogenetic inference is robust to recombination but demographic inference is not
Published this week in mBio, Jessica Hedge's new paper "Bacterial phylogenetic inference is robust to recombination but demographic inference is not" looks at a long-standing problem: why are phylogenetic trees so popular in bacterial genomics when everyone knows recombination (which is detectable in most species studied) leads to seriously misleading inference? A burst of research activity in the early 2000s showed that homologous recombination - which can result from various forms of horizontal gene transfer in bacteria - can distort phylogenetic trees and lead to false inference of positive selection and demographic growth in methods that rely on them.
In the intervening years there has been intense research in the field of population genetics into approaches that account for recombination, although the practically useful methods rely on approximations because of the inherent difficulties of learning about complex reticulated evolutionary networks that recombination generates. This has led many of my population genetics colleagues to regard - at least privately - the use of phylogenetic trees in recombining species as "bust", and the conclusions drawn from such studies as questionable. In this paper we show that this view is too simple.

In the intervening years there has been intense research in the field of population genetics into approaches that account for recombination, although the practically useful methods rely on approximations because of the inherent difficulties of learning about complex reticulated evolutionary networks that recombination generates. This has led many of my population genetics colleagues to regard - at least privately - the use of phylogenetic trees in recombining species as "bust", and the conclusions drawn from such studies as questionable. In this paper we show that this view is too simple.
Friday, 6 June 2014
Cheltenham Science Festival
 |
| Antibiotic Resistance Coconut Shy |
The game was more difficult than it looks, and just one visitor knocked off all five coconuts. We gave out NDM pens to the sixty visitors who managed to knock off three or more.
 |
| Microscope and Top Trumps |
 |
| Genome Evolution Dance Mat |
 |
| Outbreak Map |
Outbreak Map: We made an Outbreak Map to show the reach of our stall over the day, with visitors that scored highly on the coconut shy pushing in pins to show where they had travelled from. Had we been handing out germs instead of pens, we could have started outbreaks as far afield as Edinburgh, France and Spain, as well as a large cluster in Cheltenham and the surrounding counties.
Other research groups are representing the department throughout the week.
 |
| NDM Microbiology Stall at the Cheltenham Science Festival (L-R): Sarah Earle, Louise Pankhurst, Danny Wilson, Liz Batty, Dilrini De Silva, Jess Hedge, Catrin Moore. Amy Mason, Gareth Jenkin-Jones and Jane Charlesworth also helped with the preparations, and Jen Bardsley co-ordinated all the NDM Stalls. |
New paper: Mobile elements drive recombination hotspots in the core genome of Staphylococcus aureus

Compared to recent work focusing on individual, highly-related strains of S. aureus, we found much higher rates of core HGT across the species as a whole. We saw that the frequency of HGT varies along the genome. At broad scales, core HGT is higher near the origin of replication, a pattern reminiscent of the one described by Eduardo Rocha and colleagues in E. coli, who hypothesized that the over-abundance of DNA near the origin during rapid growth could promote HGT.
At fine scales, we found more frequent HGT in regions of the core genome close to mobile elements. The hottest regions occurred near mobile regions called ICE6013, SCC and genomic island α. The insertion and excision of mobile elements from the genome represents a type of HGT, so our finding that nearby core regions also experience more HGT suggests there is some sort of "spill over". This idea is supported by work in Ashley Robinson's group that found similarities between ICE6013 and a class of mobile elements in Streptococcus agalactiae called TnGBS2. TnGBS2 was discovered by Phillipe Glaser's lab who showed it sometimes transfers large tracts of adjacent core material during conjugation.
Whether conjugation alone can explain the high levels of core HGT we saw in S. aureus is unclear - our results suggest there is detectable HGT even in core regions far from mobile elements. Transformation is another possible mechanism of core HGT, but S. aureus is generally thought to be naturally incapable of transformation. However, intriguing work published by Tarek Msadek and colleagues in 2012 indicates there may be cryptic mechanisms of transformation in S. aureus after all. It remains to be seen whether the relative contributions of transformation, transduction and conjugation to the long-term evolution of S. aureus can be disentangled.
Tuesday, 1 October 2013
The role of hospital transmission in Clostridium difficile infection
 This week the Modernising Medical Microbiology consortium at Oxford published the findings of a six-year study into the transmission of the hospital "superbug" Clostridium difficile. The research, which appears in the New England Journal of Medicine, shows that the majority of new cases cannot be traced to other infections in hospital, and indicates instead that there must be a large, as yet unidentified, reservoir of C. difficile infectious to humans. This finding is important because it suggests that there is a limit to which more and more intense hospital cleaning - important though it has been - can continue to have in reducing C. difficile infection.
This week the Modernising Medical Microbiology consortium at Oxford published the findings of a six-year study into the transmission of the hospital "superbug" Clostridium difficile. The research, which appears in the New England Journal of Medicine, shows that the majority of new cases cannot be traced to other infections in hospital, and indicates instead that there must be a large, as yet unidentified, reservoir of C. difficile infectious to humans. This finding is important because it suggests that there is a limit to which more and more intense hospital cleaning - important though it has been - can continue to have in reducing C. difficile infection.The research, which is the result of a tireless effort by a large number of my colleagues - notably David Eyre, Tim Peto and Sarah Walker - used bacterial whole genome sequencing to detect within-hospital transmission by searching for extremely closely related bacterial strains among more than 1200 cases of C. difficile infection that occurred in Oxfordshire between September 2007 and March 2011. The consortium is currently developing the approach for routine microbiology diagnostics and infection control, with a view to eventual roll-out across the NHS.
Friday, 20 September 2013
Postdoctoral Position in Statistical Genomics
The position of Postdoctoral Scientist is available in my group to lead research on the Wellcome Trust and Royal Society
funded project Statistical Methods for Whole Genome Phenotype Mapping in
Bacterial Populations.
Bacteria cause disease throughout the world. Different strains vary in disease severity, but the genetic variants responsible remain largely undiscovered. Recent breakthroughs in whole genome sequencing provide new opportunities for discovery, but the lack of statistical analysis tools tailored to the special structure of bacterial populations presents a roadblock. The goal of the project is to develop an analysis framework for mapping genes underlying naturally variable traits in bacterial populations. Focusing on the hospital-associated pathogens Staphylococcus aureus and Clostridium difficile, we will investigate the role of bacterial variants on disease severity.
The role of the Postdoctoral Scientist is to develop novel statistical methods for analysing genotype-phenotype associations in bacteria at the whole genome level. The successful candidate will write software implementing the statistical methods and apply them to design and carry out investigations into the genetic basis of virulence in natural populations of bacterial pathogens. The ideal candidate would be a recently graduating PhD student with experience of statistical genetics and computer programming, with evidence of publicly released software. Experience of population genetics or microbiology would be advantageous but is not essential.
The post is available immediately, and is available for up to 3 years in the first instance. For more details on this position, including salary, job description, selection criteria and how to apply, please see the University of Oxford recruitment page.
Applications for this vacancy are to be made online. The closing date is 12.00 noon on Monday 4 November 2013. Applicants will be asked to upload a CV and a supporting statement as part of the online application. For informal enquiries, please email me. More information about the group's research is available here.
Bacteria cause disease throughout the world. Different strains vary in disease severity, but the genetic variants responsible remain largely undiscovered. Recent breakthroughs in whole genome sequencing provide new opportunities for discovery, but the lack of statistical analysis tools tailored to the special structure of bacterial populations presents a roadblock. The goal of the project is to develop an analysis framework for mapping genes underlying naturally variable traits in bacterial populations. Focusing on the hospital-associated pathogens Staphylococcus aureus and Clostridium difficile, we will investigate the role of bacterial variants on disease severity.
The role of the Postdoctoral Scientist is to develop novel statistical methods for analysing genotype-phenotype associations in bacteria at the whole genome level. The successful candidate will write software implementing the statistical methods and apply them to design and carry out investigations into the genetic basis of virulence in natural populations of bacterial pathogens. The ideal candidate would be a recently graduating PhD student with experience of statistical genetics and computer programming, with evidence of publicly released software. Experience of population genetics or microbiology would be advantageous but is not essential.
The post is available immediately, and is available for up to 3 years in the first instance. For more details on this position, including salary, job description, selection criteria and how to apply, please see the University of Oxford recruitment page.
Applications for this vacancy are to be made online. The closing date is 12.00 noon on Monday 4 November 2013. Applicants will be asked to upload a CV and a supporting statement as part of the online application. For informal enquiries, please email me. More information about the group's research is available here.
Tuesday, 17 September 2013
Sir Henry Dale Fellowship

The project addresses the question of how to detect genes or mutations in bacteria responsible for variability in important traits such as the tendency to cause human disease. Focusing on the hospital-associated pathogens Staphylococcus aureus and Clostridium difficile, the project has the potential to help identify genetic variants that explain why some bacteria cause more severe infections, knowledge that could help develop new drugs and tests that improve patient treatment.
The fellowship runs for five years, and includes support for a postdoctoral research assistant and laboratory costs. I will be advertising a position shortly. If you are interested, please get in touch.
I want to thank the funders and reviewers for supporting this project, and my colleagues who helped me write and re-write the research proposal.
Thursday, 6 June 2013
Detecting mixed strain infections with whole genome sequencing
Whole genome sequencing in near-to-real time is set to become a routine tool for outbreak detection by hospital and public health microbiology labs, following successful pilot studies in the UK last year. Typically, the bacteria are cultured from a clinical sample, and a single colony is picked for sequencing. Since a bacterial colony grows from a single cell, this procedure ensures that all the cells picked for sequencing are genetically identical, and this in turn helps piece the genome back together again following sequencing.
But it exposes the system to a flaw. What would happen if a patient sick with two strains transmitted one, but not the other to a second patient? Characterizing the genome of just one of the strains in the first patient risks missing the transmission event entirely, because the "wrong" strain might have been sequenced.
One safeguard would be to sequence multiple bacterial colonies per sample, three for example. But this would increase the cost of routine surveillance three-fold.
In a new paper published this month in PLoS Computational Biology, with David Eyre, Madeleine Cule, Sarah Walker and others, we have investigated an alternative solution, where by a large number of colonies gets sequenced all together. The cost is the same as that of sequencing a single colony. But the downstream bioinformatics analysis is complicated considerably by the presence of multiple strains. To cope with this, we developed a new computational method that reconstructs the identities of the multiple strains, using a panel of reference genomes to help where possible.
By applying the approach to 26 clinical samples of Clostridium difficile hospital infections with known epidemiological relationships, we detected four mixed strain infections, one of which revealed a previously undetected transmission event within the hospital. For full details, read the open access paper.
But it exposes the system to a flaw. What would happen if a patient sick with two strains transmitted one, but not the other to a second patient? Characterizing the genome of just one of the strains in the first patient risks missing the transmission event entirely, because the "wrong" strain might have been sequenced.
One safeguard would be to sequence multiple bacterial colonies per sample, three for example. But this would increase the cost of routine surveillance three-fold.
In a new paper published this month in PLoS Computational Biology, with David Eyre, Madeleine Cule, Sarah Walker and others, we have investigated an alternative solution, where by a large number of colonies gets sequenced all together. The cost is the same as that of sequencing a single colony. But the downstream bioinformatics analysis is complicated considerably by the presence of multiple strains. To cope with this, we developed a new computational method that reconstructs the identities of the multiple strains, using a panel of reference genomes to help where possible.
By applying the approach to 26 clinical samples of Clostridium difficile hospital infections with known epidemiological relationships, we detected four mixed strain infections, one of which revealed a previously undetected transmission event within the hospital. For full details, read the open access paper.
Wednesday, 22 May 2013
Within-host evolution of Staphylococcus aureus during asymptomatic carriage
 Given its notoriety as one of the world's major causes of infection-related deaths, it may come as a surprise that one in three healthy adults carry the human pathogen Staphylococcus aureus in their noses without adverse effects. Indeed, most people carry the bacteria at some point in their lives. So carriage must be seen as the normal state of affairs in the human-S. aureus interaction, and by understanding this state better we can improve our understanding of why, in some people, the bacteria go on to cause life-threatening invasive disease.
Given its notoriety as one of the world's major causes of infection-related deaths, it may come as a surprise that one in three healthy adults carry the human pathogen Staphylococcus aureus in their noses without adverse effects. Indeed, most people carry the bacteria at some point in their lives. So carriage must be seen as the normal state of affairs in the human-S. aureus interaction, and by understanding this state better we can improve our understanding of why, in some people, the bacteria go on to cause life-threatening invasive disease.
This month sees publication of an investigation by my colleagues and me into the evolution of S. aureus during this normal healthy carriage state. The carriers in our study harboured populations of the bacteria that were very closely related but typically not identical, implying that the bacteria had evolved within the human body. The nose appears to be a microcosm of evolution for S. aureus, showing all the different types of genetic variation known at the species level within the noses of these individual carriers. For the most part, within-host evolution of the bacteria was very conservative, but certain proteins expressed on the surface of the bacteria and toxins secreted by the bacteria showed evidence of involvement in a host-pathogen arms race.
The paper, whose lead authors include Tanya Golubchik, Liz Batty, Derrick Crook and Rory Bowden, has received coverage on the EveryONE blog and F1000. I liked Gerald Pier's conclusion, made on the post-publication peer review website: "Given that about 30% of the world's seven billion-plus humans, and an unknown number of animals, are chronically colonized with S. aureus, the tremendous opportunity provided to this organism for generating genetic variation to counteract human efforts to prevent S. aureus infections may be one of the most formidable barriers to overcome in order to develop vaccines and highly effective interventions to lessen the impact of this organism on human and animal health."
Monday, 4 February 2013
Coalescent inference for infectious disease

This paper appears as part of an issue on Next-generation molecular and evolutionary epidemiology of infectious disease, which accompanies a Royal Society discussion meeting organized by Oli Pybus, Christophe Fraser and Andrew Rambaut. The Royal Society has made audio recordings of the talks at this meeting, and the accompanying satellite meeting, available online, including my talk on Bethany's paper.
Thursday, 15 November 2012
Postdoctoral Positions in Pathogen Genomics
These positions are now closed. There are currently seven posts advertised to join the Pathogen Genomics group at the Nuffield Department of Medicine in Oxford. Prof Derrick Crook and colleagues are seeking exceptional, creative, quantitatively minded scientists to join a multidisciplinary team of researchers using population genomics to understand the evolution and transmission of human pathogens. We are seeking to appoint a number of promising young researchers to extend our existing strengths in the areas of phylogenomics, statistical genetics and bioinformatics.
The group is studying a range of bacterial and viral pathogens including tuberculosis, Staphylococcus aureus, Clostridium difficile, HIV, norovirus and hepatitis C virus. Our research interests include within-host evolution, the genetic basis of virulence, transmission dynamics and outbreak investigation via real-time genomics.
A major translational goal of the project is to exploit the transformative effect of population genomics on bacteriology to improve routine clinical practice in public health and microbiology laboratories.
The research is supported by the UKCRC Modernising Medical Microbiology Consortium, the Health Innovation Challenge Fund, the NHS National Institute for Health Research, the Oxford Biomedical Research Centre, Institut Merieux and the Oxford Martin School, and pursued in collaboration with clinical colleagues in Leeds, Birmingham and Brighton, the Health Protection Agency and the WTSI.
The deadline for applications varies by position, between 26-28 November 2012.
For examples of recent papers see:
http://www.thelancet.com/journals/laninf/article/PIIS1473-3099%2812%2970277-3/fulltext
http://www.pnas.org/content/109/12/4550.full
http://bmjopen.bmj.com/content/2/3/e001124.full.pdf+html
http://www.nature.com/nrg/journal/v13/n9/pdf/nrg3226.pdf
http://www.plospathogens.org/article/info%3Adoi%2F10.1371%2Fjournal.ppat.1002874
For more information visit:
http://www.modmedmicro.ac.uk
http://www.oxfordmartin.ox.ac.uk/projects/view/127
The group is studying a range of bacterial and viral pathogens including tuberculosis, Staphylococcus aureus, Clostridium difficile, HIV, norovirus and hepatitis C virus. Our research interests include within-host evolution, the genetic basis of virulence, transmission dynamics and outbreak investigation via real-time genomics.
A major translational goal of the project is to exploit the transformative effect of population genomics on bacteriology to improve routine clinical practice in public health and microbiology laboratories.
The research is supported by the UKCRC Modernising Medical Microbiology Consortium, the Health Innovation Challenge Fund, the NHS National Institute for Health Research, the Oxford Biomedical Research Centre, Institut Merieux and the Oxford Martin School, and pursued in collaboration with clinical colleagues in Leeds, Birmingham and Brighton, the Health Protection Agency and the WTSI.
The deadline for applications varies by position, between 26-28 November 2012.
For examples of recent papers see:
http://www.thelancet.com/journals/laninf/article/PIIS1473-3099%2812%2970277-3/fulltext
http://www.pnas.org/content/109/12/4550.full
http://bmjopen.bmj.com/content/2/3/e001124.full.pdf+html
http://www.nature.com/nrg/journal/v13/n9/pdf/nrg3226.pdf
http://www.plospathogens.org/article/info%3Adoi%2F10.1371%2Fjournal.ppat.1002874
For more information visit:
http://www.modmedmicro.ac.uk
http://www.oxfordmartin.ox.ac.uk/projects/view/127
Monday, 5 November 2012
James Martin Fellowship
This position is now closed. A prestigious James Martin Fellowship funded by the Oxford Martin School is available in my research group for a highly motivated and creative population geneticist interested in developing cutting edge methods for the analysis of high-throughput whole genome sequencing data to better understand the evolution and epidemiology of the major pathogens HIV and Hepatitis C Virus.
The position, which is part of the Curing Chronic Viral Infections project, is fully funded for three years and is affiliated with the Institute for Emerging Infections, the Modernising Medical Microbiology consortium, the Peter Medawar Building for Pathogen Research and the Nuffield Department of Medicine. The ideal candidate will have a track record in statistical or computational genetics and experience of programming in a language such as C++ or Java.
Full details can be found on the University of Oxford Recruitment website. Please send informal enquiries, with a CV, to me by email. The deadline for applications is 12 noon on 27th November 2012.
The position, which is part of the Curing Chronic Viral Infections project, is fully funded for three years and is affiliated with the Institute for Emerging Infections, the Modernising Medical Microbiology consortium, the Peter Medawar Building for Pathogen Research and the Nuffield Department of Medicine. The ideal candidate will have a track record in statistical or computational genetics and experience of programming in a language such as C++ or Java.
Full details can be found on the University of Oxford Recruitment website. Please send informal enquiries, with a CV, to me by email. The deadline for applications is 12 noon on 27th November 2012.
Friday, 7 September 2012

Thursday, 16 August 2012
gammaMap available for download

Monday, 13 August 2012
Nature Reviews Genetics: Transforming Clinical Microbiology
My colleagues Xavier Didelot, Rory Bowden, Tim Peto, Derrick Crook and I have just published a review online ahead of print in Nature Reviews Genetics called Transforming clinical microbiology with bacterial genome sequencing.
You might also be interested to read a similarly themed review recently published by our friends at the University of Cambridge and Wellcome Trust Sanger Institute in PLoS Pathogens titled Routine use of microbial whole genome sequencing in diagnostic and public health microbiology.
These review articles follow hot on the heels of a pair of research articles published by our two groups: A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance in BMJ Open and Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak in the New England Journal of Medicine. The common thread is the impact of near-to-real-time whole genome sequencing on outbreak detection and other translational activities in hospitals and public health laboratories.
You might also be interested to read a similarly themed review recently published by our friends at the University of Cambridge and Wellcome Trust Sanger Institute in PLoS Pathogens titled Routine use of microbial whole genome sequencing in diagnostic and public health microbiology.
These review articles follow hot on the heels of a pair of research articles published by our two groups: A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance in BMJ Open and Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak in the New England Journal of Medicine. The common thread is the impact of near-to-real-time whole genome sequencing on outbreak detection and other translational activities in hospitals and public health laboratories.
Friday, 20 July 2012
Post-doc Positions in Pathogen Genomics
Post-doc positions in Pathogen Genomics are available in my group and Derrick Crook's lab. We will be hiring people to work on pathogen whole genome sequence analysis and bioinformatics. More details available soon. In the meantime, find out about our research:
If you are interested, please get in touch.
If you are interested, please get in touch.
Monday, 5 March 2012
PNAS paper on staphylococcal evolution during infection
 Today in PNAS Early Edition my colleagues and I have a paper published reporting the genome evolution of Staphylococcus aureus during the transition from prolonged nasal carriage to invasive disease. Since Staph. aureus, a major bacterial cause of life-threatening infections, is carried without symptoms by a quarter of healthy adults, a natural question is to ask what genetic changes - if any - accompany the transition to invasive disease. The opportunity to pursue this question arose from a detailed epidemiological investigation of asymptomatic Staph. aureus nasal carriage set up by colleagues of mine including Derrick Crook and Kyle Knox. The study has recruited over 1,000 participants in Oxfordshire since it began running in October 2008. One participant developed a bloodstream infection that was indistinguishable from the strain of Staph. aureus persistently carried in the nose for the previous 13 months. Members of the Modernising Medical Microbiology consortium, led by Derrick and Rory Bowden, sequenced the genomes of 68 bacterial colonies isolated from the nasal and blood samples from this participant, and 101 colonies from nasal samples from two other participants that did not go on to develop disease. Bernadette Young and Tanya Golubchik analyzed the genome evolution of these bacterial populations, discovering an unusual pattern in the mutations that occurred between nasal carriage and invasive disease: mutations that led to prematurely truncated proteins were significantly over-represented, including one in a gene previously associated with virulence in bacteria. To know more, read the full open access article.
Today in PNAS Early Edition my colleagues and I have a paper published reporting the genome evolution of Staphylococcus aureus during the transition from prolonged nasal carriage to invasive disease. Since Staph. aureus, a major bacterial cause of life-threatening infections, is carried without symptoms by a quarter of healthy adults, a natural question is to ask what genetic changes - if any - accompany the transition to invasive disease. The opportunity to pursue this question arose from a detailed epidemiological investigation of asymptomatic Staph. aureus nasal carriage set up by colleagues of mine including Derrick Crook and Kyle Knox. The study has recruited over 1,000 participants in Oxfordshire since it began running in October 2008. One participant developed a bloodstream infection that was indistinguishable from the strain of Staph. aureus persistently carried in the nose for the previous 13 months. Members of the Modernising Medical Microbiology consortium, led by Derrick and Rory Bowden, sequenced the genomes of 68 bacterial colonies isolated from the nasal and blood samples from this participant, and 101 colonies from nasal samples from two other participants that did not go on to develop disease. Bernadette Young and Tanya Golubchik analyzed the genome evolution of these bacterial populations, discovering an unusual pattern in the mutations that occurred between nasal carriage and invasive disease: mutations that led to prematurely truncated proteins were significantly over-represented, including one in a gene previously associated with virulence in bacteria. To know more, read the full open access article.
Wednesday, 11 January 2012
SMBE 2012: Microbial Genome Evolution Symposium

High-throughput sequencing makes it possible for the first time to sequence hundreds of microbial genomes rapidly at low cost. These methods have huge potential to significantly improve our understanding of microbial evolution, so that many research projects have recently been set up to generate and analyze such data. This symposium will provide an overview of the progress made by such projects, as well as the many challenges they pose. It is now possible to identify the vast majority of SNPs within large population samples of microbial isolates. These datasets are illuminating the molecular, ecological and population-level dynamic processes occurring over short time scales in natural populations inhabiting a range of habitats from the clinic to the environment. We aim to explore these recent advances and the development of new methods of analyses required to fully exploit these extremely large sequence datasets. Relevant topics include quantifying the variation in the rates of recombination and mutation between closely related lineages, the evolution of base composition, the relative power of drift and selection, examining the acquisition of adaptive traits (e.g. antibiotic resistance, host adaptation, metabolic flexibility, regulatory changes) within a phylogenetic framework, and the distribution of variation over time and space (phylogeography). The role of phage and conjugative elements in structuring populations as both vehicles for gene flow and parasitic elements will also be considered. The symposium will focus on variation within natural populations rather than experimental evolution.
Friday, 2 December 2011
New method inferring natural selection published today
Saturday, 1 January 2011
Group Member Profiles Updated

Richard is investigating patterns of genetic diversity and linkage disequilibrium in Staphylococcus aureus, while Bethany is studying the transmission dynamics of norovirus using population genetics and epidemiological modelling. Both are funded jointly by the UKCRC project Modernising Medical Microbiology and the Nuffield Department of Clinical Medicine. For more information, see their individual profiles.
Friday, 1 October 2010
Geographical differences in transmission revealed by cryptic population structure
Two papers that I co-authored with colleagues at Lancaster and Massey Universities appear this month in the October 2010 issue of Epidemiology & Infection. The common theme is that cryptic differences in the population structure of the enteric pathogen Campylobacter jejuni, revealed by my method for attributing cases to source populations, suggest subtle differences in transmission between rural and urban districts.
 The method, implemented in the software iSource (available on my website), allows strains of campylobacter to be characterized as poultry- or cattle-associated based on their genetic profiles. Interestingly, when the relative incidence of poultry- and cattle-associated strains is plotted on a map, there is a significantly higher occurrence of poultry-related disease in urban areas and cattle-related disease in rural areas. Both studies – one in Lancashire led by Edith Gabriel and one in New Zealand led by Petra Mullner – draw the same conclusion. These findings imply that there are subtle differences in transmission in rural and urban areas. Whether they represent geographical differences in the profile of food pathogens, environmental exposure, resistance to infection or other risk factors is not understood.
The method, implemented in the software iSource (available on my website), allows strains of campylobacter to be characterized as poultry- or cattle-associated based on their genetic profiles. Interestingly, when the relative incidence of poultry- and cattle-associated strains is plotted on a map, there is a significantly higher occurrence of poultry-related disease in urban areas and cattle-related disease in rural areas. Both studies – one in Lancashire led by Edith Gabriel and one in New Zealand led by Petra Mullner – draw the same conclusion. These findings imply that there are subtle differences in transmission in rural and urban areas. Whether they represent geographical differences in the profile of food pathogens, environmental exposure, resistance to infection or other risk factors is not understood.

Saturday, 18 September 2010
Evolutionary Genetics for Translational Research

- Tracking the transmission of hospital-acquired pathogens
- Understanding transmission dynamics at the population level
- Identifying the mechanistic and adaptive basis of disease
- Explaining how pathogens emerge, persist and spread globally
Saturday, 10 July 2010
What are the conditions for multiple foci of adaptation?
Selection on standing variation, soft sweeps, parallel adaptation: these alternatives to the population genetics paradigm of the S-shaped selective sweep have in common the idea that the response of a species to a change in selection pressure may frequently involve multiple mutations, which may arise in multiple locales, and which may appear at different sites in the genome. Consequently, the footprint of selection in the genome is different to that expected under a single selective sweep and therefore likely to be missed by scans of the genome looking for selection.
Many examples of parallel adaptation have been put forward, for instance multiple drug resistance in the malaria parasite Plasmodium vivax. But how plausible is parallel adaptation as an evolutionary mechanism, and what are the conditions that make it likely? These questions were addressed by Graham Coop presenting joint work with his postdoc Peter Ralph in one of the stand-out talks of the SMBE conference in Lyon.
Their key finding is that the multifarious parameters that go into building a spatial model of adaptation (strength of selection, the mutation rate, population density, average dispersal distance of offspring) can be distilled down to a single key quantity: the characteristic length given by the equation
 When the geographical extent of the species range exceeds this characteristic length, the conditions are right for parallel adaptation. Graham's talk made accessible the complex mathematics behind this result. He has kindly made the slides available (click here) and the paper is now available at the Genetics website (click here).
When the geographical extent of the species range exceeds this characteristic length, the conditions are right for parallel adaptation. Graham's talk made accessible the complex mathematics behind this result. He has kindly made the slides available (click here) and the paper is now available at the Genetics website (click here).
Many examples of parallel adaptation have been put forward, for instance multiple drug resistance in the malaria parasite Plasmodium vivax. But how plausible is parallel adaptation as an evolutionary mechanism, and what are the conditions that make it likely? These questions were addressed by Graham Coop presenting joint work with his postdoc Peter Ralph in one of the stand-out talks of the SMBE conference in Lyon.
Their key finding is that the multifarious parameters that go into building a spatial model of adaptation (strength of selection, the mutation rate, population density, average dispersal distance of offspring) can be distilled down to a single key quantity: the characteristic length given by the equation

Subscribe to:
Posts (Atom)








{kind=link}